


发布时间:2022-11-25 来源:瀚海量子

图1 获ACM Gordon Bell奖提名
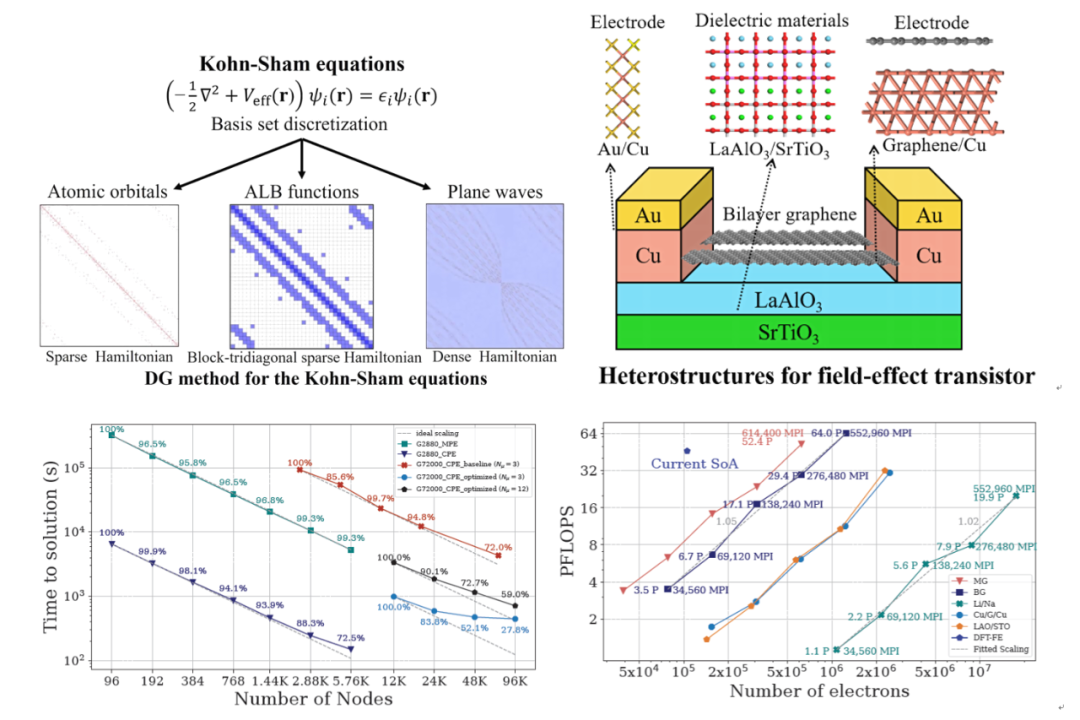

第一性原理(first-principles)电子结构计算的主要任务就是从量子力学基本原理出发,在无任何经验参数的条件下通过现代计算机从头计算分子、固体和纳米材料等的物理化学性质及其应用。该研究方法不需要开展真实的实验,极大地节省了实验成本,缩短了新材料的开发周期,为材料的制备和改性、新材料的开发以及极端环境下材料的性质研究提供了有效的理论指导。因此,第一性原理电子结构计算是二十一世纪解决实验理论问题和预测新材料结构性能的强有力工具和标准研究方法。
合肥瀚海量子专业从事基于量子力学的第一性原理电子结构计算和分子动力学模拟等软件研发,以中国科学技术大学杨金龙院士课题组胡伟研究团队负责运营推广的多款软件产品为核心产品,致力于打破国内计算软件市场被国外软件垄断的僵局。瀚海量子的软件采用了目前国际上最先进的低标度数值计算方法和迭代对角化算法,结合软件硬件一体化高性能并行计算设计,尤其是针对国产超级计算机的异构并行框架,优于本领域其它所有计算软件,是目前做第一性原理固体分子材料计算体系最大(最高250万原子)、效率最高(支持GPU-CUDA加速,并全面适配国产超算)的计算软件,可赋能新材料、新能源、生物制药、人工智能、量子力学科普科教等多个领域。



量子大会邀请函
